


 Sign Out
Sign Out

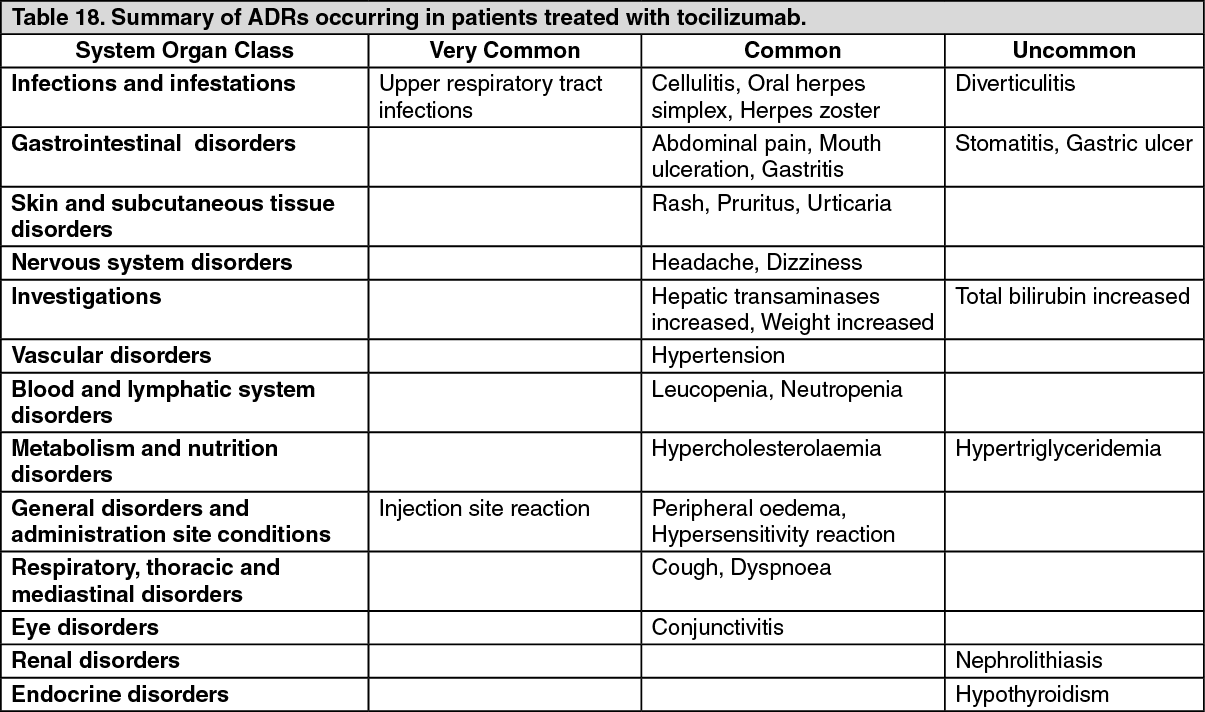
Adverse Drug Reactions (ADRs) from clinical trials (Table 18) are listed by MedDRA system organ class according to clinical importance to the patient. The corresponding frequency category for each ADR is based on the following convention: very common (≥1/10), common (≥1/100 to < 1/10) or uncommon (≥1/1000 to < 1/100). (See Table 18.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/imageDescription of selected adverse drug reactions from clinical trials: Rheumatoid Arthritis: Patients Treated with Intravenous Tocilizumab: The safety of tocilizumab has been studied in 5 Phase III, double-blind controlled trials and their extension periods.
The all control population includes all patients from the double-blind phases of each core study from randomization until either the first change in the treatment regimen, or two years is reached. The control period in 4 of the studies was 6 months and in 1 study was up to 2 years. In the double-blind controlled studies, 774 patients received tocilizumab 4 mg/kg in combination with MTX, 1870 patients received tocilizumab 8 mg/kg in combination with MTX/other DMARDs, and 288 patients received tocilizumab 8 mg/kg monotherapy.
The all exposure population includes all patients who received at least one dose of tocilizumab either in the double-blind control period or open label extension phase in studies. Of the 4009 patients in this population, 3577 received treatment for at least 6 months, 3296 for at least one year; 2806 received treatment for at least 2 years and 1222 for 3 years.
Infections: In the 6 month controlled trials, the rate of all infections reported with tocilizumab 8 mg/kg+DMARD treatment was 127 events per 100 patient (pt) years compared to 112 events per 100 pt years in the placebo+DMARD group. In the all exposure population the overall rate of infections with tocilizumab was 108 events per 100 pt years exposure.
In 6 month controlled clinical trials rate of serious infections (bacterial, viral and fungal) with tocilizumab 8 mg/kg+DMARD was 5.3 events per 100 pt years exposure compared to 3.9 events per 100 pt years exposure in the placebo+DMARD group. In the monotherapy study the rate of serious infections was 3.6 events per 100 pt years of exposure in the tocilizumab group and 1.5 events per 100 pt years of exposure in the MTX group.
In the all exposure population the overall rate of serious infections was 4.7 events per 100 pt years. Reported serious infections, some with fatal outcome, included pneumonia, cellulitis, herpes zoster, gastroenteritis, diverticulitis, sepsis, bacterial arthritis. Cases of opportunistic infections have also been reported.
Gastrointestinal Perforation: During the 6 month controlled clinical trials, the overall rate of gastrointestinal perforation was 0.26 events per 100 pt years with tocilizumab therapy. In the all exposure population the overall rate of gastrointestinal perforation was 0.28 events per 100 pt years. Reports of gastrointestinal perforation on tocilizumab were primarily reported as complications of diverticulitis including generalized purulent peritonitis, lower GI perforation, fistula and abscess.
Infusion Reactions: In the 6 month controlled trials adverse events associated with infusion (selected events occurring during or within 24 hours of infusion) were reported by 6.9% of patients in the tocilizumab 8 mg/kg+DMARD and 5.1% of patients in the placebo+DMARD group. Events reported during the infusion were primarily episodes of hypertension; events reported within 24 hours of finishing an infusion were headache and skin reactions (rash, urticaria). These events were not treatment limiting.
The rate of anaphylaxis (occurring in a total of 6/3778 patients) was several-fold higher in the 4 mg/kg arm in comparison to the 8 mg/kg dose. Clinically significant hypersensitivity reactions associated with tocilizumab and requiring treatment discontinuation, were reported in a total of 13 out of 3778 patients (0.3%) treated with tocilizumab during the controlled and open label clinical trials. These reactions were generally observed during the second to fifth infusions of tocilizumab (see General under Precautions).
Immunogenicity: A total of 2876 patients have been tested for anti-tocilizumab antibodies in the 6 month controlled clinical trials. Forty six patients (1.6%) developed positive anti-tocilizumab antibodies of whom 5 had an associated medically significant hypersensitivity reaction leading to withdrawal. Thirty patients (1.1%) developed neutralizing antibodies.
Early Rheumatoid Arthritis: Study VI (WA19926) evaluated 1162 patients with early, moderate to severe RA who were naïve to treatment with both MTX and a biologic agent. The overall safety profile observed in the tocilizumab treatment groups was consistent with the known safety profile of tocilizumab (see Table 18) (see Pharmacology: Pharmacodynamics: Clinical/Efficacy Studies under Actions).
Monotherapy: tocilizumab versus adalimumab: In a 24 week double-blinded, parallel study (monotherapy with tocilizumab 8 mg/kg IV q4w (N=162) compared to adalimumab 40 mg SC q2w (N=162)), the overall clinical adverse event profile was similar between tocilizumab and adalimumab. The proportion of patients with serious adverse events was balanced between the treatment groups (tocilizumab 11.7% vs. adalimumab 9.9%) with the most common event being infections (3.1% each). Both study treatments induced the same pattern of changes in laboratory safety parameters (decreases in neutrophil and platelet counts, increases in ALT, AST and lipids), however, the magnitude of change and the frequency of marked abnormalities was higher with tocilizumab compared with adalimumab. Four (2.5%) patients in the tocilizumab arm and two (1.2%) patients in the adalimumab arm experienced CTC grade 3 or 4 neutrophil count decreases. Eleven (6.8%) patients in the tocilizumab arm and five (3.1%) patients in the adalimumab arm experienced ALT increases of CTC grade 2 or higher. The mean LDL increase from baseline was 0.64 mmol/l (25 mg/dL) for patients in the tocilizumab arm and 0.19 mmol/L (7 mg/dL) for patients in the adalimumab arm. The safety observed in the tocilizumab arm was consistent with the known safety profile of tocilizumab and no new or unexpected adverse drug reactions were observed (see Table 18) (see Pharmacology: Pharmacodynamics: Clinical/Efficacy Studies under Actions).
Patients Treated with Subcutaneous Tocilizumab: The safety of subcutaneous tocilizumab in RA was studied in SC-I. The study compared the efficacy and safety of tocilizumab 162 mg administered every week SC versus 8 mg/kg IV in 1262 subjects with adult RA. All patients in the study received background non-biologic DMARD(s). The safety and immunogenicity observed for tocilizumab administered SC was consistent with the known safety profile of IV tocilizumab and no new or unexpected adverse drug reactions were observed (see Table 18). A higher frequency of injection site reactions (ISRs) was observed in the SC arms compared with placebo SC injections in the IV arms (see Pharmacology: Pharmacodynamics: Clinical/Efficacy Studies under Actions).
Injection Site Reactions (ISRs): During the 6-month controlled period, in SC-I, the frequency of ISRs was 10.1% (64/631) and 2.4% (15/631) for the SC tocilizumab and the SC placebo (IV group) weekly injections, respectively. These ISRs (including erythema, pruritus, pain and haematoma) were mild to moderate in severity. The majority was resolved without any treatment and none necessitated drug discontinuation.
Immunogenicity: In SC-I, a total of 625 patients treated with tocilizumab 162 mg weekly were tested for anti-tocilizumab antibodies in the 6 month controlled period. Five patients (0.8%) developed positive anti-tocilizumab antibodies; of these, all developed neutralizing anti-tocilizumab antibodies.
A total of 1454 SC tocilizumab all exposure patients have been tested for anti-tocilizumab antibodies, thirteen patients (0.9%) developed positive anti-tocilizumab antibodies, and of these 12 patients (0.8%) developed neutralizing anti-tocilizumab antibodies.
No correlation of antibody development to clinical response or adverse events was observed.
Giant Cell Arteritis: The safety of subcutaneous tocilizumab has been studied in one Phase III study (WA28119) with 251 GCA patients. The total patient years duration in the tocilizumab all exposure population was 138.5 patient years during the 12-month double blind, placebo-controlled phase of the study. The overall safety profile observed in the tocilizumab treatment groups was consistent with the known safety profile of tocilizumab (see Table 18) (see Pharmacology: Pharmacodynamics: Clinical/Efficacy Studies under Actions).
Infections: The rate of infection/serious infection events was balanced between the tocilizumab weekly group (200.2/9.7 events per 100 patient years) versus placebo plus 26 weeks prednisone taper (156.0/4.2 events per 100 patient years) and placebo plus 52 weeks taper (210.2/12.5 events per 100 patient years) groups.
Polyarticular Juvenile Idiopathic Arthritis: The safety profile of tocilizumab was studied in 240 paediatric patients with pJIA. In Study WA19977, 188 patients (2 to 17 years of age) were treated with IV tocilizumab and in Study WA28117, 52 patients (1 to 17 years of age) were treated with SC tocilizumab. The total patient exposure to tocilizumab in the pJIA all exposure population was 184.4 patient years for IV tocilizumab and 50.4 patient years for SC tocilizumab. In general, the safety profile observed in patients with pJIA was consistent with the known safety profile of tocilizumab with the exception of ISRs (see Table 18). A higher frequency of ISRs was experienced by pJIA patients following SC tocilizumab injections compared to adult RA patients (see as previously mentioned).
Infections: Infections are the most commonly observed events in pJIA. The rate of infections in the pJIA IV tocilizumab all exposure population was 163.7 per 100 patient years. The most common events observed were nasopharyngitis and upper respiratory tract infections. The rate of serious infections was numerically higher in patients weighing below 30 kg treated with 10 mg/kg tocilizumab (12.2 per 100 patient years) compared to patients weighing ≥30 kg, treated with 8 mg/kg tocilizumab (4.0 per 100 patient years). The incidence of infections leading to dose interruptions was also numerically higher in patients weighing below 30 kg treated with 10 mg/kg tocilizumab (21.4%) compared to patients weighing ≥30 kg, treated with 8 mg/kg tocilizumab (7.6%). The rate of infection in pJIA patients treated with SC tocilizumab was comparable with pJIA patients treated with IV tocilizumab.
Infusion Reactions: In pJIA patients, infusion related reactions are defined as all events occurring during or within 24 hours of an infusion with IV tocilizumab. In the tocilizumab all exposure population, 11 patients (5.9%) experienced infusion reactions during the infusion, and 38 patients (20.2%) experienced an event within 24 hours of an infusion. The most common events occurring during infusion were headache, nausea and hypotension and within 24 hours of infusion were dizziness and hypotension. In general, the adverse drug reactions observed during or within 24 hours of an infusion were similar in nature to those seen in RA and sJIA patients (see as previously mentioned).
No clinically significant hypersensitivity reactions associated with tocilizumab and requiring treatment discontinuation were reported.
Injection Site Reactions: A total of 28.8% (15/52) pJIA patients experienced ISRs to SC tocilizumab. These ISRs occurred in 44% of patients ≥30 kg compared to 14.8% of patients below 30 kg. The most common ISRs were injection site erythema, swelling, hematoma, pain and pruritis. All ISRs reported were non-serious Grade 1 events, and none of the ISRs required patient withdrawal from treatment or dose interruption.
Immunogenicity: Across the two studies in pJIA patients, a total of four patients (0.5% [1/188] in the IV Study WA19977 and 5.8% [3/52] in the SC Study WA28117) developed positive neutralizing anti-tocilizumab antibodies without developing a serious or clinically significant hypersensitivity reaction. Of these 4 patients, 2 subsequently withdrew from the study. No correlation between antibody development and clinical response or adverse events was observed.
Systemic Juvenile Idiopathic Arthritis: The safety profile of tocilizumab in sJIA was studied in 163 paediatric patients. In Study WA18221 (12-week trial and long term extension), 112 patients (2 to 17 years of age) were treated with IV tocilizumab and in Study WA28118 (52-week trial), 51 patients (1 to 17 years of age) were treated with SC tocilizumab. In general, the adverse drug reactions in patients with sJIA were similar in type to those seen in RA patients (see as previously mentioned).
Infections: In the 12 week controlled trial (Study WA18221), the rate of all infections in the IV tocilizumab group was 344.7 per 100 patient-years and 287.0 per 100 patient-years in the placebo group. In the open label extension study (Part II) the overall rate of infections remained similar at 306.6 per 100 patient- years.
In the 12 week controlled trial (Study WA18221), the rate of serious infections in the IV tocilizumab group was 11.5 per 100 patient years. In the open label extension study the overall rate of serious infections remained stable at 11.3 per 100 patient years. Reported serious infections were similar to those seen in RA patients with the addition of varicella and otitis media.
The rate of infection in sJIA patients treated with SC tocilizumab was comparable to sJIA patients treated with IV tocilizumab.
Infusion Reactions: For sJIA patients, infusion related reactions are defined as all events occurring during or within 24 hours of an infusion with IV tocilizumab. In the 12 week controlled trial (Study WA18221), four percent (4.0%) of patients from the tocilizumab group experienced events occurring during infusion, one event (angioedema) was considered serious and life-threatening, and the patient was discontinued from study treatment.
In the 12 week controlled trial experience, 16% of patients in the IV tocilizumab group and 5.4% of patients in the placebo group experienced an event within 24 hours of infusion. In the tocilizumab group, the events included, but not limited to rash, urticaria, diarrhoea, epigastric discomfort, arthralgia and headache. One of these events, (urticaria) was considered serious.
Clinically significant hypersensitivity reactions associated with IV tocilizumab and requiring treatment discontinuation, were reported in 1 out of 112 patients (below 1%) treated with IV tocilizumab during the controlled and open-label parts of the clinical trial.
Injection Site Reactions (ISRs): In Study WA28118, a total of 41.2% (21/51) sJIA patients experienced ISRs to SC tocilizumab. The most common ISRs were erythema, pruritus, pain, and swelling at the injection site. The majority of ISRs reported were Grade 1 events and all ISRs reported were non-serious and none of the ISRs required patient withdrawal from treatment or dose interruption.
Immunogenicity: In Study WA18221, all 112 patients were tested for anti-tocilizumab antibodies at baseline. Two patients developed positive anti-tocilizumab antibodies with one of these patients having a hypersensitivity reaction leading to withdrawal. In Study WA28118, 46 of the 51 (90.2%) patients tested for anti-tocilizumab antibodies at baseline had at least one post-baseline screening assay result. No patient developed positive anti-tocilizumab antibodies post-baseline.
Laboratory Abnormalities: Haematology abnormalities: Neutrophils: There was no clear relationship between decreases in neutrophils below 1 x 109/L and the occurrence of serious infections in any of the indications.
Rheumatoid Arthritis: Intravenous Administration: In the 6 month controlled trials decreases in neutrophil counts below 1 x 109/L occurred in 3.4% of patients on tocilizumab 8 mg/kg+DMARD compared to below 0.1% of patients on placebo+DMARD. Approximately half of the instances of ANC below 1 x 109/L occurred within 8 weeks of starting therapy. Decreases below 0.5 x 109/L were reported in 0.3% patients receiving tocilizumab 8 mg/kg +DMARD (see Dosage & Administration and General under Precautions).
In the all control and all exposure population, the pattern and incidence of decreases in neutrophil counts remained consistent with what was seen in the 6 month controlled clinical trials.
Subcutaneous Administration: During routine laboratory monitoring in the tocilizumab 6-month controlled period of clinical trial SC-I, a decrease in neutrophil count below 1 × 109/L occurred in 2.9% of patients on tocilizumab 162 mg SC weekly.
Giant Cell Arteritis: During routine laboratory monitoring in the tocilizumab 12-month double blind, placebo-controlled phase of study WA28119, a decrease in neutrophil count below 1 x 109/L occurred in 4% of patients in the tocilizumab SC weekly group. This was not observed in either of the placebo plus prednisone taper groups.
Polyarticular Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the tocilizumab all exposure population, a decrease in neutrophil count below 1 × 109/L occurred in 3.7% of patients treated with IV tocilizumab and 15.4% of patients treated with SC tocilizumab.
Systemic Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the 12-week controlled trial (Study WA18221), a decrease in neutrophil counts below 1 × 109/L occurred in 7% of patients in the IV tocilizumab group, and in none in the placebo group.
In the open-label extension study (WA18221), decreases in neutrophil counts below 1 x 109/L, occurred in 15% of the IV tocilizumab group.
In the 52-week open-label trial (Study WA28118), neutrophil count decrease below 1 × 109/L occurred in 23.5% of patients treated with SC tocilizumab.
Platelets: Rheumatoid Arthritis: Intravenous Administration: In the 6 month controlled trials decreases in platelet counts below 100 x 103/μL occurred in 1.7% of patients on tocilizumab 8 mg/kg plus traditional DMARDs compared to below 1% of patients on placebo plus traditional DMARDs, without associated bleeding events (see Dosage & Administration and General under Precautions).
In the all control and all exposure population, the pattern and incidence of decreases in platelet counts remained consistent with what was seen in the 6 month controlled clinical trials.
Subcutaneous Administration: During routine laboratory monitoring in the tocilizumab 6-month controlled period of clinical trial SC-I, none of the patients had a decrease in platelet count to ≤50 × 103/μL.
Giant Cell Arteritis: During routine laboratory monitoring in the tocilizumab 12-month double blind, placebo-controlled phase of study WA28119, one patient (1%, 1/100) in the tocilizumab SC weekly group had a single transient occurrence of decreased platelet count below 100 x 103/μL without associated bleeding events. A decrease in platelet count below 100 x 103 / μL was not observed in either of the placebo plus prednisone taper groups.
Polyarticular Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the tocilizumab all exposure population, a decrease in platelet count to ≤50 × 103/μL occurred in 1% patients treated with IV tocilizumab, without associated bleeding events and in no patients treated with SC tocilizumab.
Systemic Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the 12-week controlled trial (Study WA18221), 3% of patients in the placebo group and 1% in the IV tocilizumab group had a decrease in platelet count to ≤ 100 × 103/μL.
In the open-label extension study (WA18221), decreases in platelet counts below 100 x 103/μL occurred in 3% of patients of the IV tocilizumab group, without associated bleeding events.
In the 52-week open-label trial (Study WA28118), decreases in platelet counts below 100 × 103/μL occurred in 2% of patients treated with SC tocilizumab.
Liver enzyme elevations: Rheumatoid Arthritis: Intravenous Administration: During the 6 month controlled trials transient elevations in ALT/AST above 3xULN were observed in 2.1% of patients on tocilizumab 8 mg/kg compared to 4.9% of patients on MTX, and in 6.5% of patients who received tocilizumab 8 mg/kg + DMARD compared to 1.5% of patients on placebo+DMARD. The addition of potentially hepatotoxic drugs (e.g. MTX) to tocilizumab monotherapy resulted in increased frequency of these elevations. Elevations of ALT/AST above 5xULN were observed in 0.7% of tocilizumab monotherapy patients and 1.4% of tocilizumab+DMARD patients, the majority of whom were discontinued from tocilizumab treatment (see Dosage & Administration and General under Precautions). During routine laboratory monitoring, the incidence of indirect bilirubin greater than the upper limit of normal was 6.2% in patients treated with 8 mg/kg tocilizumab + DMARD in the all control population.
In the all control and all exposure population, the pattern and incidence of elevations in ALT/AST remained consistent with what was seen in the 6 month controlled clinical trials.
In Study VI, MTX-naïve adult patients with moderate to severe, active early RA (mean disease duration ≤ 6 months) experienced more transient elevations in ALT above 3xULN compared with the all control population. This was observed in both tocilizumab treated patients and MTX monotherapy patients.
In Study WA25204, of the 1538 patients with moderate to severe RA (see Pharmacology: Pharmacodynamics: Clinical/Efficacy Studies under Actions) and treated with tocilizumab, elevations in ALT or AST >3 x ULN occurred in 5.3% and 2.2% patients, respectively. One serious event of drug induced hepatitis with hyperbilirubinemia was reported in association with tocilizumab treatment (see General under Precautions).
Subcutaneous Administration: During routine laboratory monitoring in the tocilizumab 6-month controlled period of clinical trial SC-I, elevation in ALT or AST ≥3 x ULN occurred in 6.5% and 1.4% of patients, respectively on SC weekly.
Giant Cell Arteritis: During routine laboratory monitoring in the tocilizumab 12-month double blind, placebo-controlled phase of study WA28119, elevation in ALT ≥3 ULN occurred in 3% of patients in the tocilizumab SC weekly group compared to 2% in the placebo plus 52 week prednisone taper group and none in the placebo plus 26 weeks prednisone taper group. An elevation in AST > 3 ULN occurred in 1% of patients in the tocilizumab SC weekly group, compared to no patients in either of the placebo plus prednisone taper group.
Polyarticular Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the tocilizumab all exposure population, elevation in ALT or AST ≥3 x ULN occurred in 3.7% and below 1% of patients treated with IV tocilizumab, and in 9.6% and 3.8% patients treated with SC tocilizumab, respectively.
Systemic Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the 12-week controlled trial (Study WA18221), elevation in ALT or AST ≥ 3xULN occurred in 5% and 3% of patients, respectively, in the IV tocilizumab group, and in 0% of placebo patients.
In the open-label extension study (WA18221), elevation in ALT or AST ≥ 3xULN occurred in 12% and 4% of patients, respectively, in the IV tocilizumab group.
In the 52-week open-label trial (Study WA28118), elevation in ALT or AST ≥3 x ULN occurred in 9.8% and 4.0% patients treated with SC tocilizumab, respectively.
Elevations in lipid parameters: Rheumatoid Arthritis: Intravenous Administration: During routine laboratory monitoring in the 6 month controlled trials, elevations in lipid parameters (total cholesterol, LDL, HDL, triglycerides) were observed in patients treated with tocilizumab. Approximately 24% of patients receiving tocilizumab in clinical trials experienced sustained elevations in total cholesterol above 6.2 mmol/L (240 mg/dL), with 15% experiencing a sustained increase in LDL to ≥ 4.1 mmol/L (160 mg/dL).
In the majority of patients there was no increase in atherogenic indices, and elevations in total cholesterol responded to treatment with lipid-lowering agents.
In the all control and all exposure population, the pattern and incidence of elevations in lipid parameters remained consistent with what was seen in the 6 month controlled clinical trials.
Subcutaneous Administration: During routine laboratory monitoring in the tocilizumab 6-month controlled period of clinical trial SC-I, 19% of patients on SC weekly experienced sustained elevations in total cholesterol above 6.2 mmol/L (240 mg/dL), with 9% experiencing a sustained increase in LDL to ≥ 4.1 mmol/L (160 mg/dL) on SC weekly.
Giant Cell Arteritis: During routine laboratory monitoring in the tocilizumab 12-month double blind, placebo-controlled phase of study WA28119, 29% of patients experienced elevations in total cholesterol above 6.2 mmol/L (240 mg/dL), with 12% experiencing an increase in LDL to ≥ 4.1 mmol/L (160 mg/dL) in the tocilizumab SC weekly group.
Polyarticular Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the IV tocilizumab Study WA19977 3.4% and 10.4% of patients experienced a post-baseline elevation of their LDL-cholesterol value to ≥ 130 mg/dL and total cholesterol value to ≥ 200 mg/dL at any time during the study treatment, respectively. In the SC tocilizumab Study WA28117, 14.3% and 12.8% of patients experienced a post-baseline elevation of their LDL-cholesterol value to ≥ 130 mg/dL and total cholesterol value to ≥ 200 mg/dL at any time during study treatment, respectively.
Systemic Juvenile Idiopathic Arthritis: During routine laboratory monitoring in the 12-week controlled trial (Study WA18221), 13.4% and 33.3% of patients experienced a post-baseline elevation of their LDL-cholesterol value to ≥ 130 mg/dL and total cholesterol value to ≥ 200 mg/dL, respectively.
In the open-label extension study (WA18221), 13.2% and 27.7% of patients experienced a post-baseline elevation of their LDL-cholesterol value to ≥ 130 mg/dL and total cholesterol value to ≥ 200 mg/dL, respectively.
In the 52-week open-label trial (Study WA28118), 23.4% and 35.4% of patients experienced a post-baseline elevation of their LDL-cholesterol value to ≥ 130 mg/dL and total cholesterol value to ≥ 200 mg/dL, respectively.
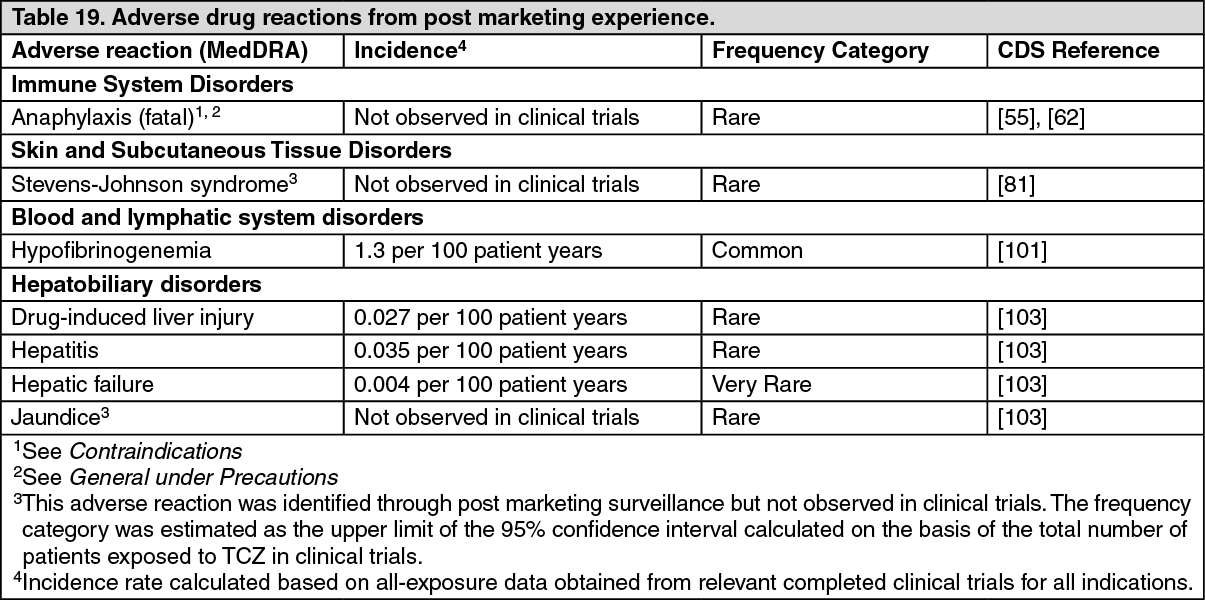
Post Marketing Experience: The following adverse drug reactions have been identified from post marketing experience with tocilizumab (Table 19) based on spontaneous case reports, literature cases and cases from non-interventional study programs. Adverse drug reactions are listed according to system organ classes in MedDRA and the corresponding frequency category estimation for each adverse drug reaction is based on the following convention: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000). (See Table 19.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
View ADR Monitoring Form